
China’s fast-growing medical device market presents numerous opportunities for European companies.  However, understanding the practicalities of accessing the market is not simple. In order to enter Chinese market, all medical devices are required to obtain a pre-market approval, known as medical device registration, from the Chinese Food and Drug Administration (CFDA). In the following article the EU SME Centre looks at the regulations pertaining to medical devices in China and describe some of the more notable aspects.
However, understanding the practicalities of accessing the market is not simple. In order to enter Chinese market, all medical devices are required to obtain a pre-market approval, known as medical device registration, from the Chinese Food and Drug Administration (CFDA). In the following article the EU SME Centre looks at the regulations pertaining to medical devices in China and describe some of the more notable aspects.
The Regulation for Supervision and Administration of Medical Devices (Regulation) is currently the highest level of legislation in China’s medical devices sector. The latest version has been in effect since 1st June, 2014, which, in comparison to the previous version, has nearly doubled in length and changed significantly in terms of scope.
It is therefore advisable that European SMEs doing business in China’s medical device sector to take a close look at the Regulation and seek professional advice before taking any concrete steps to enter the Chinese market.
Definition of medical devices
The definition of medical devices—based on the ‘purpose’ of use—has been amended and expanded in the Regulation. Previously, the definition included the following purposes:
- Prevention, diagnosis, treatment, monitoring or remission of diseases.
- Diagnosis, treatment, monitoring, remission or compensation of injury or physical disability.
- Research, replacement or adjustment of an anatomical or physiological process.
- Control of pregnancy.
The new regulation adds an additional two categories: “support or maintenance of life” and “samples taken from humans to provide information for medical treatment or diagnostic purposes”.
Classification
Medical devices in China are categorised into three different classes – Class I, II and III, with risk levels ranging from low to high. The criteria for classification is based on the purpose of use, the structural features of the device, whether the device has direct contact with the human body and the methods and status of use.
This classification is used predominantly for risk management where different rules are adopted:
- Class I: low level of risk, safety and effectiveness can be ensured through routine administration.
- Class II: medium level of risk, further control is required to ensure their safety and effectiveness.
- Class III: high levels of risk, special measures with strict control and administration must be enforced to achieve safety and effectiveness.
Even though this classification system may appear similar to the European system, European applicants are still advised to carefully check the Chinese classification list published by the CFDA, which is the foundation of the registration requirement and procedures.
Device registration
The new regulation eliminates the pre-marketing registration requirement for Class I devices, but not for Class II and Class III devices, which still need to register through a licensing procedure. Class I devices only need to undergo a ‘filing’ procedure with a municipal regulator (i.e., below the provincial level).
The application procedure and timeframe for medical device registration can be seen below:
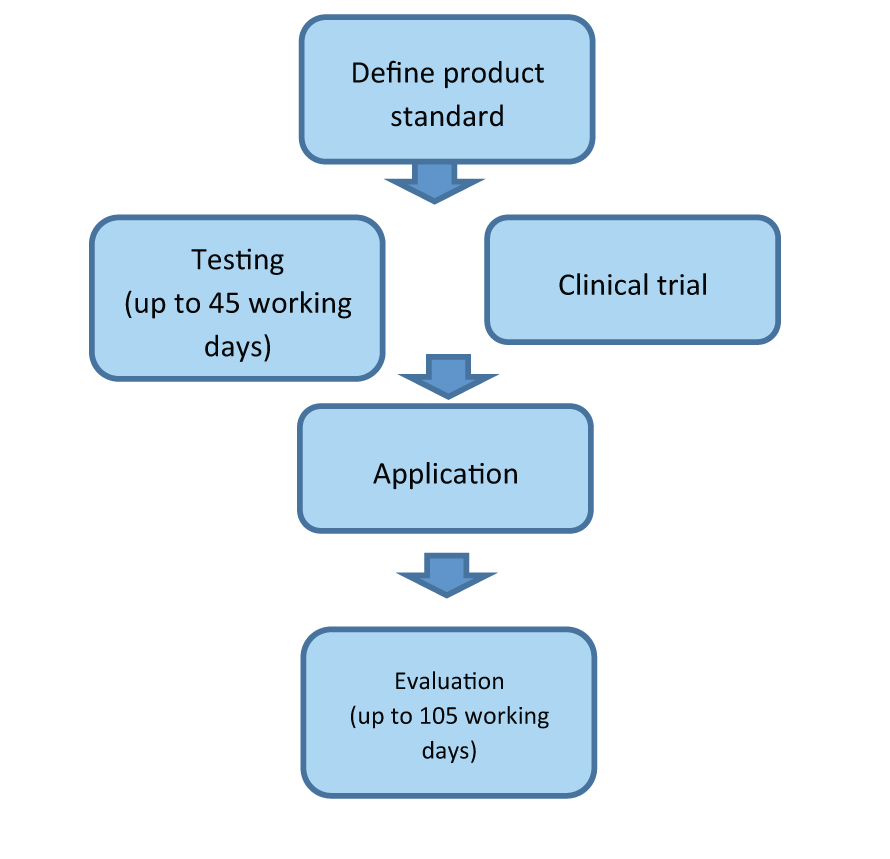
The SFDA point out on its website that the application procedure can take 105 working days, however, this includes the time period for testing or conducting a clinical trial. Overall, the process can be divided into five steps:
- Appointing an agent – the agent must be a legal Chinese entity.
- Standards and testing – compliance testing must take place at a designated laboratory in China.
- Clinical trial – depending on approval and classification a clinical trial may be required.
- Application – application and supporting documentation must be submitted.
- Evaluation – the Centre for Medical Device Evaluation (CDME) conducts a technical evaluation of the information provided then issues a registration.
Clinical Trials
With respect to clinical trials, significant clarifications and modifications have been made in the Regulation. Class I devices are no longer required to have China-based trials prior to registration, whereas most Class II and Class III devices must successfully undergo China-based clinical trials before registration and approval for marketing can be given.
The Regulation clarifies that some of the Class II and Class III devices can be exempted from the clinical trial requirement. You can find the complete list on CFDA’s website: http://www.sda.gov.cn/WS01/CL0087/105224.html.
Distribution
The notification and licensing requirements for entities distributing Class I and II devices have also been simplified. When distributing Class I devices, going through a filing procedure with the municipal-level food and drug regulatory authority is no longer required, however, distributors of Class II devices still need to follow this step. While Class II device distributors are released from obligation of obtaining a distribution licence, which sometimes can be a lengthy procedure, this does not apply to distributors of Class III devices.
The Regulation also adds a requirement for keeping records of sales, which applies to wholesalers of Class II and Class III devices as well as retailers of Class III devices.
Further resources
More comprehensive information on medical device regulations in China can be found at www.eusmecentre.org.cn. If you have questions specific to your products, please contact our experts at www.eusmecentre.org.cn/expert.
The EU SME Centre in Beijing provides a comprehensive range of hands-on support services to European small and medium-sized enterprises (SMEs), getting them ready to do business in China.
Our team of experts provides advice and support in four areas – business development, law, standards and conformity and human resources. Collaborating with external experts worldwide, the Centre converts valuable knowledge and experience into practical business tools and services easily accessible online. From first-line advice to in-depth technical solutions, we offer services through Knowledge Centre, Advice Centre, Training Centre, SME Advocacy Platform and Hot-Desks.
The Centre is an initiative implemented with the financial support of the European Union. It is managed by a consortium of six partners – the China-Britain Business Council, the Benelux Chamber of Commerce, the China-Italy Chamber of Commerce, the French Chamber of Commerce in China, the EUROCHAMBRES, and the European Union Chamber of Commerce in China.
To learn more about the Centre, visit website www.eusmecentre.org.cn

Recent Comments